Tutorials_ST_HE_downstream
[1]:
import os
os.chdir("/public/home/off_liukunpeng/project/11_cluster_problem")
import scanpy as sc
import pandas as pd
import matplotlib.pyplot as plt
[6]:
AgaeSMO__adata=sc.read_h5ad("AgaeSMO/result/Slide_1_.h5ad")
[7]:
annotation=pd.read_csv("../7_public_data/Data/osfstorage-archive/slide1_annotation.csv")
annotation
AgaeSMO__adata.obs["GrounTruth"]=list(annotation["annotation"])
[8]:
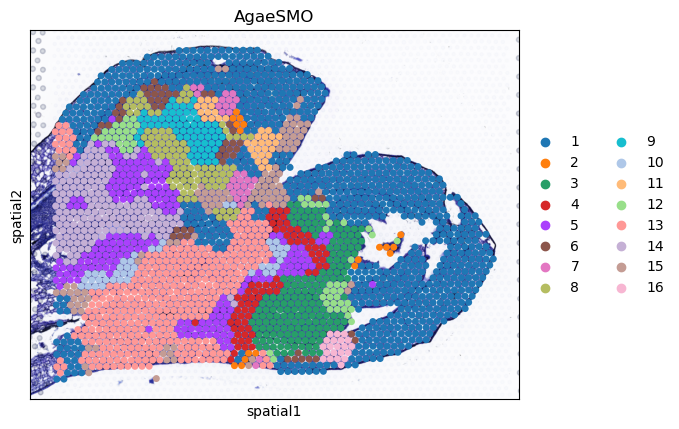
sc.pl.spatial(AgaeSMO__adata, basis='spatial', color='AgaeSMO', title='AgaeSMO', s=12, show=False)
/public/home/off_liukunpeng/software/anaconda3/envs/pyg1/lib/python3.9/site-packages/scanpy/plotting/_tools/scatterplots.py:1234: FutureWarning: The default value of 'ignore' for the `na_action` parameter in pandas.Categorical.map is deprecated and will be changed to 'None' in a future version. Please set na_action to the desired value to avoid seeing this warning
color_vector = pd.Categorical(values.map(color_map))
[8]:
[<Axes: title={'center': 'AgaeSMO'}, xlabel='spatial1', ylabel='spatial2'>]
[9]:
x,y=9000,16000
crop_coord=(x,x+5000,y,y+2000)
fig,ax=plt.subplots(2,1,figsize=(5.5,4))
sc.pl.spatial(AgaeSMO__adata[[i in [3,4,5] for i in AgaeSMO__adata.obs["AgaeSMO"]],:],
basis='spatial', color='AgaeSMO', title='AgaeSMO', s=10, show=False,ax=ax[0],
crop_coord=crop_coord)
sc.pl.spatial(AgaeSMO__adata, basis='spatial', color=None, title='HE', s=0, show=False,ax=ax[1],
crop_coord=crop_coord)
plt.subplots_adjust(left=0.05, bottom=0.05, right=0.95, top=0.95, wspace=0.05, hspace=0.2)
fig.savefig("plot/fig5_orchid_he.png",dpi=600)
/public/home/off_liukunpeng/software/anaconda3/envs/pyg1/lib/python3.9/site-packages/scanpy/plotting/_tools/scatterplots.py:1234: FutureWarning: The default value of 'ignore' for the `na_action` parameter in pandas.Categorical.map is deprecated and will be changed to 'None' in a future version. Please set na_action to the desired value to avoid seeing this warning
color_vector = pd.Categorical(values.map(color_map))

[10]:
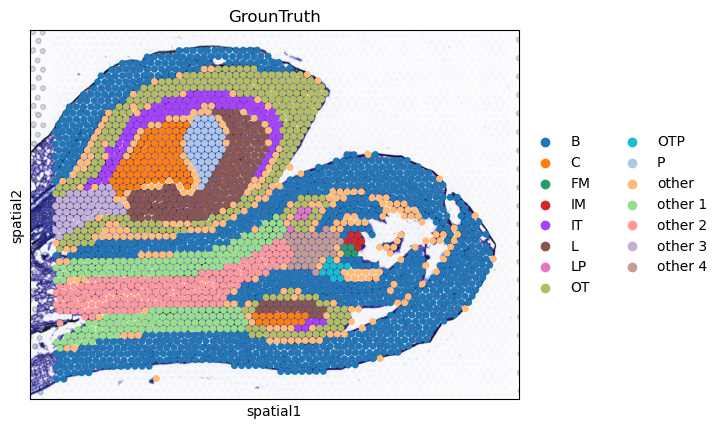
sc.pl.spatial(AgaeSMO__adata, basis='spatial', color='GrounTruth', title='GrounTruth', s=12, show=False)
/public/home/off_liukunpeng/software/anaconda3/envs/pyg1/lib/python3.9/site-packages/scanpy/plotting/_tools/scatterplots.py:1234: FutureWarning: The default value of 'ignore' for the `na_action` parameter in pandas.Categorical.map is deprecated and will be changed to 'None' in a future version. Please set na_action to the desired value to avoid seeing this warning
color_vector = pd.Categorical(values.map(color_map))
[10]:
[<Axes: title={'center': 'GrounTruth'}, xlabel='spatial1', ylabel='spatial2'>]
[11]:
import seaborn as sns
def plot_weight_value(alpha, label, modality1='mRNA', modality2='protein',show=True):
"""\
Plotting weight values
"""
import pandas as pd
df = pd.DataFrame(columns=[modality1, modality2, 'label'])
df[modality1], df[modality2] = alpha[:, 0], alpha[:, 1]
df['label'] = label
df = df.set_index('label').stack().reset_index()
df.columns = ['label', 'Modality', 'Weight value']
return df
[13]:
df_all=[]
for n,i in enumerate(set(AgaeSMO__adata.obs["AgaeSMO"])):
alpha=AgaeSMO__adata.obsm["alpha"][AgaeSMO__adata.obs["AgaeSMO"]==i,:]
# print(alpha.shape)
df=plot_weight_value(alpha,"AgaeSMO",modality1='mRNA', modality2='H&E')
df["cluster"]=i
df_all.append(df)
df_all = pd.concat(df_all, ignore_index=True)
[15]:
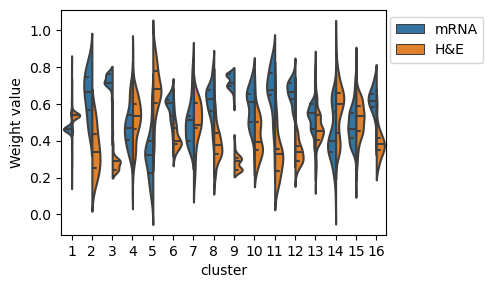
fig,ax=plt.subplots(1,figsize=(5,3))
sns.violinplot(data=df_all,ax=ax, x="cluster", y="Weight value", hue="Modality", split=True, inner="quart")
plt.subplots_adjust(left=0.15, bottom=0.15, right=0.8, top=0.90, wspace=0.05, hspace=0.05)
ax.legend( ncol=1,loc= 'upper right' ,fontsize=10,bbox_to_anchor=(1.32, 1))
[15]:
<matplotlib.legend.Legend at 0x2ae1c2256e80>
[16]:
# fig,ax=plt.subplots(2,3,figsize=(9,3))
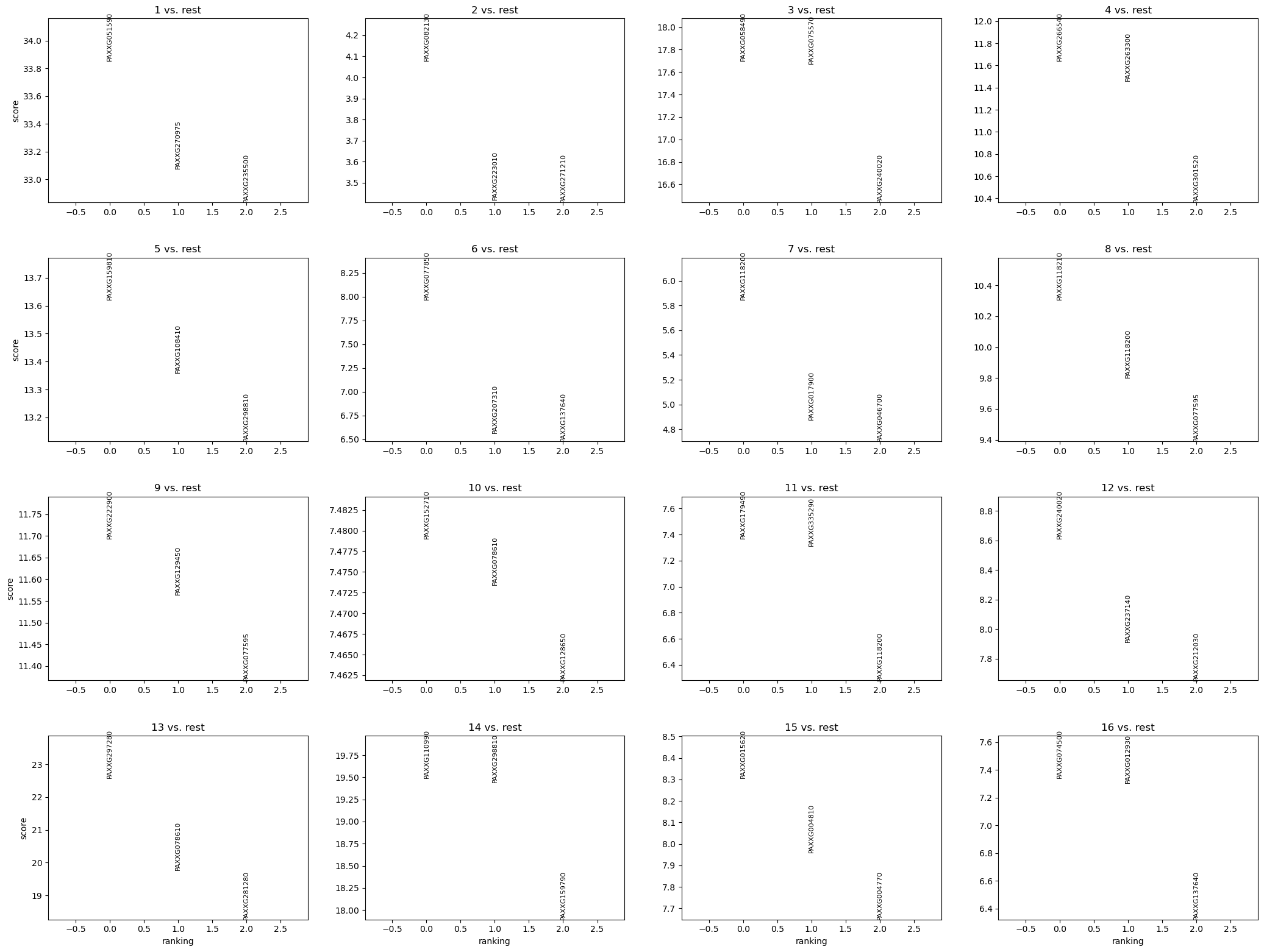
sc.tl.rank_genes_groups(AgaeSMO__adata, "AgaeSMO", method="wilcoxon")
# sc.pl.rank_genes_groups(adata, n_genes=25, sharey=False)
# sc.pl.rank_genes_groups(adata, n_genes=25,groups="2",ax=ax[0,1],sharey=False,show=False)
# sc.pl.rank_genes_groups(adata, n_genes=25,groups=["2"],sharey=False)
sc.pl.rank_genes_groups(AgaeSMO__adata, n_genes=3,sharey=False,show=False)
/public/home/off_liukunpeng/software/anaconda3/envs/pyg1/lib/python3.9/site-packages/numpy/core/fromnumeric.py:86: FutureWarning: The behavior of DataFrame.sum with axis=None is deprecated, in a future version this will reduce over both axes and return a scalar. To retain the old behavior, pass axis=0 (or do not pass axis)
return reduction(axis=axis, out=out, **passkwargs)
/public/home/off_liukunpeng/software/anaconda3/envs/pyg1/lib/python3.9/site-packages/scanpy/tools/_rank_genes_groups.py:422: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, 'logfoldchanges'] = np.log2(
/public/home/off_liukunpeng/software/anaconda3/envs/pyg1/lib/python3.9/site-packages/scanpy/tools/_rank_genes_groups.py:422: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, 'logfoldchanges'] = np.log2(
/public/home/off_liukunpeng/software/anaconda3/envs/pyg1/lib/python3.9/site-packages/scanpy/tools/_rank_genes_groups.py:422: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, 'logfoldchanges'] = np.log2(
/public/home/off_liukunpeng/software/anaconda3/envs/pyg1/lib/python3.9/site-packages/scanpy/tools/_rank_genes_groups.py:422: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, 'logfoldchanges'] = np.log2(
/public/home/off_liukunpeng/software/anaconda3/envs/pyg1/lib/python3.9/site-packages/scanpy/tools/_rank_genes_groups.py:422: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, 'logfoldchanges'] = np.log2(
/public/home/off_liukunpeng/software/anaconda3/envs/pyg1/lib/python3.9/site-packages/scanpy/tools/_rank_genes_groups.py:422: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, 'logfoldchanges'] = np.log2(
/public/home/off_liukunpeng/software/anaconda3/envs/pyg1/lib/python3.9/site-packages/scanpy/tools/_rank_genes_groups.py:422: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, 'logfoldchanges'] = np.log2(
/public/home/off_liukunpeng/software/anaconda3/envs/pyg1/lib/python3.9/site-packages/scanpy/tools/_rank_genes_groups.py:422: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, 'logfoldchanges'] = np.log2(
/public/home/off_liukunpeng/software/anaconda3/envs/pyg1/lib/python3.9/site-packages/scanpy/tools/_rank_genes_groups.py:422: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, 'logfoldchanges'] = np.log2(
/public/home/off_liukunpeng/software/anaconda3/envs/pyg1/lib/python3.9/site-packages/scanpy/tools/_rank_genes_groups.py:422: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, 'logfoldchanges'] = np.log2(
/public/home/off_liukunpeng/software/anaconda3/envs/pyg1/lib/python3.9/site-packages/scanpy/tools/_rank_genes_groups.py:422: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, 'logfoldchanges'] = np.log2(
/public/home/off_liukunpeng/software/anaconda3/envs/pyg1/lib/python3.9/site-packages/scanpy/tools/_rank_genes_groups.py:422: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, 'logfoldchanges'] = np.log2(
/public/home/off_liukunpeng/software/anaconda3/envs/pyg1/lib/python3.9/site-packages/scanpy/tools/_rank_genes_groups.py:422: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, 'logfoldchanges'] = np.log2(
/public/home/off_liukunpeng/software/anaconda3/envs/pyg1/lib/python3.9/site-packages/scanpy/tools/_rank_genes_groups.py:422: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, 'logfoldchanges'] = np.log2(
/public/home/off_liukunpeng/software/anaconda3/envs/pyg1/lib/python3.9/site-packages/scanpy/tools/_rank_genes_groups.py:422: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, 'logfoldchanges'] = np.log2(
/public/home/off_liukunpeng/software/anaconda3/envs/pyg1/lib/python3.9/site-packages/scanpy/tools/_rank_genes_groups.py:422: RuntimeWarning: invalid value encountered in log2
self.stats[group_name, 'logfoldchanges'] = np.log2(
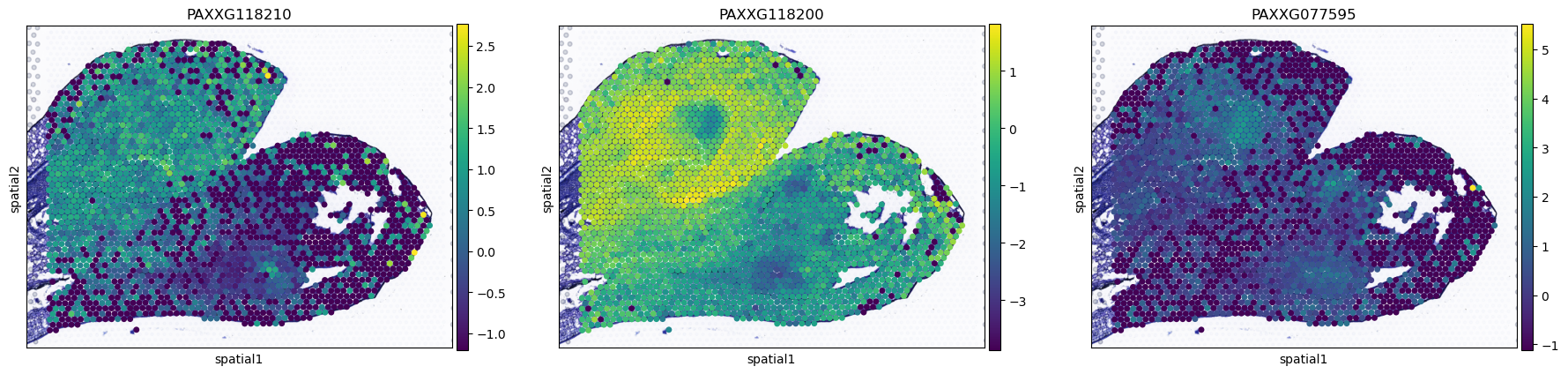
[18]:
plot_gene=sc.get.rank_genes_groups_df(AgaeSMO__adata,group="8")["names"][0:3].tolist()
sc.pl.spatial(AgaeSMO__adata, basis='spatial', color=plot_gene, s=12, show=False)
/public/home/off_liukunpeng/software/anaconda3/envs/pyg1/lib/python3.9/site-packages/scanpy/get/get.py:69: FutureWarning: The previous implementation of stack is deprecated and will be removed in a future version of pandas. See the What's New notes for pandas 2.1.0 for details. Specify future_stack=True to adopt the new implementation and silence this warning.
d = d.stack(level=1).reset_index()
[18]:
[<Axes: title={'center': 'PAXXG118210'}, xlabel='spatial1', ylabel='spatial2'>,
<Axes: title={'center': 'PAXXG118200'}, xlabel='spatial1', ylabel='spatial2'>,
<Axes: title={'center': 'PAXXG077595'}, xlabel='spatial1', ylabel='spatial2'>]
[19]:
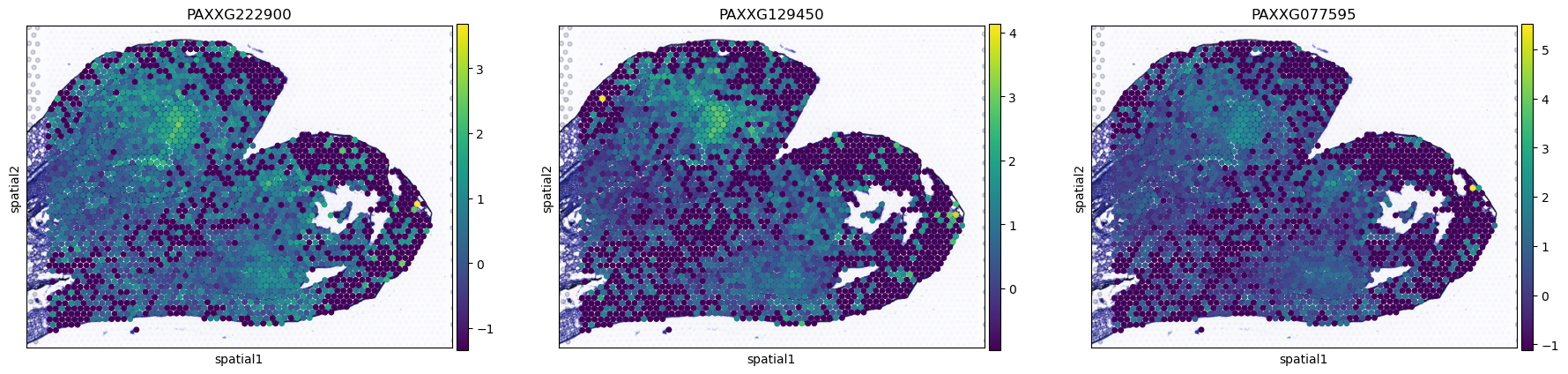
plot_gene=sc.get.rank_genes_groups_df(AgaeSMO__adata,group="9")["names"][0:3].tolist()
sc.pl.spatial(AgaeSMO__adata, basis='spatial', color=plot_gene, s=12, show=False)
/public/home/off_liukunpeng/software/anaconda3/envs/pyg1/lib/python3.9/site-packages/scanpy/get/get.py:69: FutureWarning: The previous implementation of stack is deprecated and will be removed in a future version of pandas. See the What's New notes for pandas 2.1.0 for details. Specify future_stack=True to adopt the new implementation and silence this warning.
d = d.stack(level=1).reset_index()
[19]:
[<Axes: title={'center': 'PAXXG222900'}, xlabel='spatial1', ylabel='spatial2'>,
<Axes: title={'center': 'PAXXG129450'}, xlabel='spatial1', ylabel='spatial2'>,
<Axes: title={'center': 'PAXXG077595'}, xlabel='spatial1', ylabel='spatial2'>]
[20]:
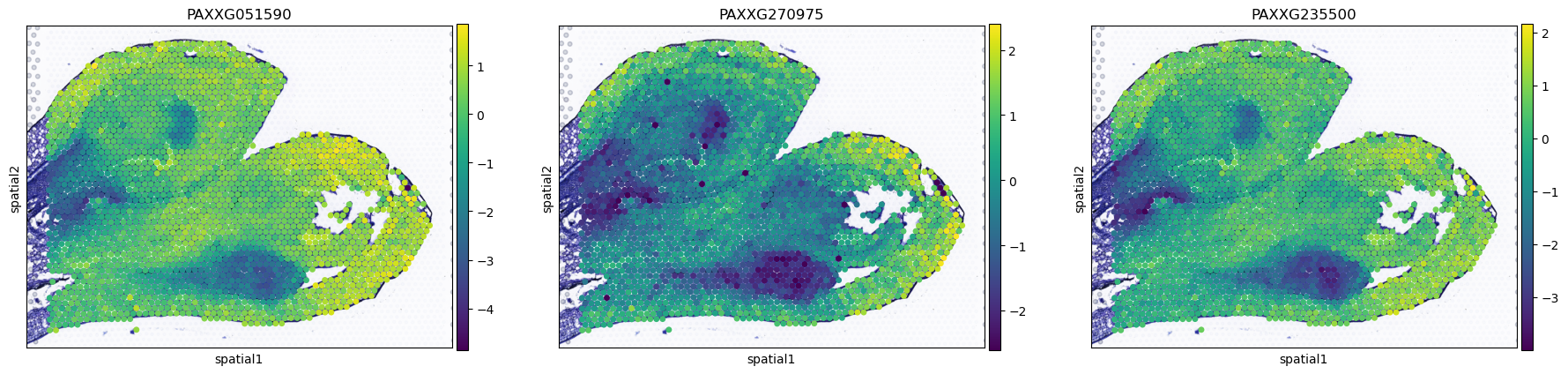
plot_gene=sc.get.rank_genes_groups_df(AgaeSMO__adata,group="1")["names"][0:3].tolist()
sc.pl.spatial(AgaeSMO__adata, basis='spatial', color=plot_gene, s=12, show=False)
/public/home/off_liukunpeng/software/anaconda3/envs/pyg1/lib/python3.9/site-packages/scanpy/get/get.py:69: FutureWarning: The previous implementation of stack is deprecated and will be removed in a future version of pandas. See the What's New notes for pandas 2.1.0 for details. Specify future_stack=True to adopt the new implementation and silence this warning.
d = d.stack(level=1).reset_index()
[20]:
[<Axes: title={'center': 'PAXXG051590'}, xlabel='spatial1', ylabel='spatial2'>,
<Axes: title={'center': 'PAXXG270975'}, xlabel='spatial1', ylabel='spatial2'>,
<Axes: title={'center': 'PAXXG235500'}, xlabel='spatial1', ylabel='spatial2'>]
[21]:
import numpy as np
import matplotlib
AgaeSMO__adata.uns['iroot'] = np.flatnonzero(AgaeSMO__adata.obs["AgaeSMO"] == 1)[0]
# sc.pp.neighbors(AgaeSMO__adata, use_rep='AgaeSMO', key_added="AgaeSMO",n_neighbors=30)
sc.pp.neighbors(AgaeSMO__adata,use_rep='AgaeSMO')
sc.tl.diffmap(AgaeSMO__adata)
sc.tl.dpt(AgaeSMO__adata)
pSM_values = AgaeSMO__adata.obs['dpt_pseudotime'].to_numpy()
# Ploting figures
matplotlib.rcParams['font.size'] = 8.0
# fig, axes = plt.subplots(1, 1, figsize=(3,3))
sz = 10
x = np.array(AgaeSMO__adata.obsm['spatial'][:,0])
y = np.array(AgaeSMO__adata.obsm['spatial'][:,1])
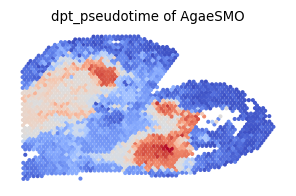
[22]:
fig, axes = plt.subplots(1, 1, figsize=(3,2))
ax_temp = axes
im = ax_temp.scatter(x, y, s=15, c=pSM_values, marker='.', cmap='coolwarm',alpha = 1)
ax_temp.axis('off')
ax_temp.set_title('dpt_pseudotime of AgaeSMO')
plt.tight_layout()
ax_temp.invert_yaxis()
# fig
[23]:
from scipy.stats import pearsonr
prs=[]
pvalue=[]
for i in range(AgaeSMO__adata.X.shape[1]):
correlation_coefficient, p_value = pearsonr(AgaeSMO__adata.X[:,i], pSM_values)
prs.append(correlation_coefficient)
pvalue.append(p_value)
[24]:
AgaeSMO__adata.var["pSM_corr"]=prs
AgaeSMO__adata.var["pSM_corr_pvalue"]=pvalue
[25]:
pSM_corr_gene=AgaeSMO__adata.var.loc[AgaeSMO__adata.var.loc[:,"pSM_corr_pvalue"]<0.05,:].sort_values("pSM_corr")
# pSM_corr_gene.loc[:,"name"]
head_gene=pSM_corr_gene.index[0:3]
pSM_corr_gene=AgaeSMO__adata.var.loc[AgaeSMO__adata.var.loc[:,"pSM_corr_pvalue"]<0.05,:].sort_values("pSM_corr",ascending=False)
# pSM_corr_gene.loc[:,"name"]
tail_gene=pSM_corr_gene.index[0:3]
[26]:
pSM_corr_gene.index
[26]:
Index(['PAXXG058490', 'PAXXG134230', 'PAXXG199840', 'PAXXG077595',
'PAXXG074880', 'PAXXG103340', 'PAXXG098390', 'PAXXG298620',
'PAXXG104000', 'PAXXG346300',
...
'PAXXG024270', 'PAXXG250980', 'PAXXG235510', 'PAXXG380090',
'PAXXG225190', 'PAXXG225210', 'PAXXG235500', 'PAXXG051590',
'PAXXG271210', 'PAXXG270975'],
dtype='object', length=13131)
[27]:
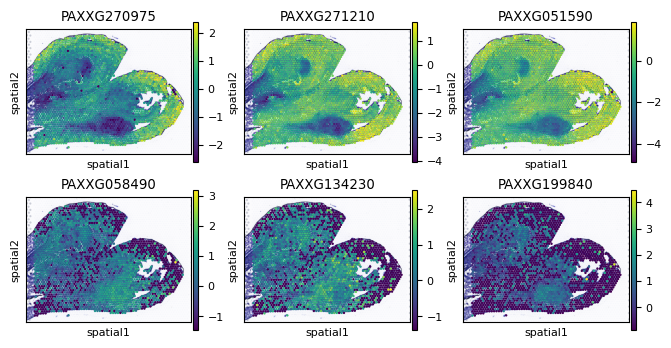
print(list(head_gene))
print(list(tail_gene))
plot_gene=list(head_gene)+list(tail_gene)
['PAXXG270975', 'PAXXG271210', 'PAXXG051590']
['PAXXG058490', 'PAXXG134230', 'PAXXG199840']
[28]:
fig, axes = plt.subplots(2, 3, figsize=(8,4))
# sc.pl.spatial(AgaeSMO__adata, basis='spatial', color=plot_gene, s=12, show=False)
for n,i in enumerate(head_gene):
sc.pl.spatial(AgaeSMO__adata, basis='spatial', color=i, ax=axes[0,n], s=12, show=False)
for n,i in enumerate(tail_gene):
sc.pl.spatial(AgaeSMO__adata, basis='spatial', color=i, ax=axes[1,n], s=12, show=False)
[ ]: